Externe Links
OMIM 300843 – Bornholm-Augenkrankheit
OMIM 310460 – x-chromosomale hohe Kurzsichtigkeit
Weitere Eigenschaften
- Die Netzhaut hat ein normales Aussehen;
- stabiler Zustand, der von Geburt an vorliegt. Einige Progressionen wurden berichtet;
- anomales oder kein photopisches ERG;
- Mutation der OPN1LW/OPN1MW-Gene.
Geschichte
Die Geschichte dieser Krankheit erweist sich aufgrund der Schwierigkeit, ihre genetische Herkunft hervorzuheben, als schwierig. Aktuelle gilt die Annahme, dass diese Krankheit durch eine seltene Mutation verursacht wird, die aus einer Reihe von Punktmutationen (Single Nucleotide Polymorphisms, kurz SNP) auf einem der 6 Exonen der Opsin-Gene (Exon 3) besteht. Diese Art von Mutation kann zu einem „Spleißdefekt“ der Opsine (Proteine) führen, was wiederum zu einer Zapfen-Dysfunktion und Farbwahrnehmungstörungen führen kann. Es ist heute nicht klar, wie Kurzsichtigkeit aus dieser Mutation resultiert, obwohl Kurzsichtigkeit ein häufiges Merkmal der Bornholm-Augenkrankheit und Blauzapfen-Monochromasie ist.
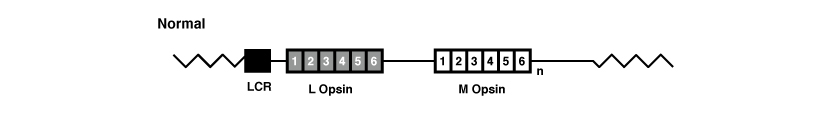
Opsin-Gen-Array
Ursprünglich in einer großen Familie der Insel Bornholm in Dänemark mit Deuteranopie und später in mehreren Familien mit Protanopie wird eine erste Art dieser Zapfendysfunktion beschrieben, die mit Verlust von Funktionsmutationen im Zapfen-Opsin-Array einhergeht und Bornholm-Krankheit (Bornholm Eye Disease, kurz BED) genannt wird.
Haim et al. (1988) [2] und [3] beschrieben eine große dänische Familie der Insel Bornholm mit x-chromosomaler Kurzsichtigkeit in Kombination mit Astigmatismus, Sehstörungen und Deuteranopie. Symptome waren bei allen betroffenen Männern vorhanden. Kurzsichtigkeit wurde im Alter von 1,5 bis 5 Jahren diagnostiziert. Das Syndrom wurde in 5 Generationen der Familie zurückverfolgt.
Young et al. (2004) [8] berichtete von einer Familie in Minnesota dänischer Herkunft mit einem ähnlichen x-chromosomalen Phänotyp. Der einzige Unterschied zwischen der Familie aus Minnesota und der ursprünglichen BED-Familie war die Farbsinnstörung. Während die betroffenen Mitglieder der BED-Familie Deuteranopie aufwiesen, konnte bei Betroffenen der Familie aus Minnesota Protanopie festgestellt werden. Bis zu diesem Punkt war nicht klar, ob es sich um eine neue Form der x-chromosomalen Krankheit mit hoher Kurzsichtigkeit und nicht-progressiver Zapfenfunktionsstörung handelte und ob diese mit Störungen der Farbwahrnehmung verbunden war oder nicht.
Bei Bartsocas et al. [1] findet sich nämlich ein Nachweis einer Familie mit x-chromosomaler hoher Kurzsichtigkeit ohne weitere Symptome von Farbblindheit. Die Krankheit ist dem Chromosom Xq28 zugeordnet und weist den ersten bezeichneten Locus hoher Kurzsichtigkeit auf (MYP1).
Hier bestimmten Hanna et al. (1997) [6] die Existenz eines Gens namens CHROMOSOM X OPEN READING FRAME 2 (CXORF2) oder TEX28-Gen, das 5 Exonen enthält. Die Transkription erfolgt in die GCP und TKTL1 entgegengesetzte Richtung. Durch Genomsequenzierung kartierten Hanna et al. (1997) [6] das TEX28-Gen zwischen den GCP- und TKTL1-Genen auf Chromosom Xq28. Sie stellten fest, dass TEX28 innerhalb des Sehpigment-Gen-Arrays auf Chromosom Xq28 mehrfach wiederholt wird, wobei jedoch der Ausschluss von Exon 1 aus den Gen-Kopien darauf hindeutete, dass die Transkription auf die Kopie zwischen den GCP- und TKTL1-Genen beschränkt ist.

(TESTIS-EXPRESSED GENE 28; TEX28 Alternative titles; symbols CHROMOSOME X OPEN READING FRAME 2; CXORF2 HGNC Approved Gene Symbol: TEX28)
Hanna et al. (1997) [6] schlugen vor, dass ich aus der Deletion der Sehpigmentgene ergebende Störungen der Farbwahrnehmung, auf mögliche assoziierte Phänotypen neu bewertet werden sollten, die sich aus einer Störung des TEX28-Gens ergeben könnten.
Chen et al. (2006) [10] stellten fest, dass auf Chromosom Xq28 3 exakte Kopien des TEX28-Gens vorhanden sind.
Metlapally et al. (2009) [11] untersuchten die Kopienzahlvariante (CNV) des CXORF2/TEX28-Gens in 5 Familien, in der die x-chromosomale starke Kurzsichtigkeit mit Zapfenfunktionsstörung abgesondert wurde, und stellten fest, dass betroffene Personen aus 4 der Familien entweder eine höhere (4 oder 5) oder niedrigere (1) Anzahl von Kopien von CXORF2/TEX28 hatten als die 3 Kopien bei nicht betroffenen Personen. Metlapally et al. (2009) [11] deuteten darauf hin, dass CXORF2/TEX28-Gen-CNVs mit x-chromosomaler Kurzsichtigkeit und dem Phänotyp der Zapfendysfunktion in Zusammenhang stehen.
Ratnamala et al. (2011) [12] beschrieben zwei große asiatisch-indische Stammbäume mit isolierter, nicht-syndromischer Kurzsichtigkeit, bei der sich die Erkrankung als x-chromosomales rezessives Merkmal abzusondern schien. Der Grad der Kurzsichtigkeit war in beiden Familien variabel und reichte von -6 bis -23 D (Mittelwert -8,48 D). Nur Männer waren betroffen. Auftritt im Alter von 4 bis 12 Jahren. Bei keinem der beiden Stammbäume wurden andere damit verbundene Sehanomalien, insbesondere Farbwahrnehmungsstörungen, gemeldet.
Aus früheren Forschungsarbeiten scheinen CXORF2/TEX28-Gen-CNVs mit dem x-chromosomalen Phänotyp mit hoher Kurzsichtigkeit + Zapfendysfunktion assoziiert zu sein, auch ohne Farbwahrnehmungsstörungen.
Am anderen Ende beschrieben in denselben Jahren Michaelides et al.(2005) [9] vier britische nicht-konsanguine Familien mit einem x-chromosomalen Zapfendysfunktionssyndrom, das mit Kurzsichtigkeit verbunden ist, ähnlich der von Young et al.(2004)[8] beschriebenen Erkrankung in der Familie in Minnesota. Die betroffenen Mitglieder hatten eine mittlere bis hohe Kurzsichtigkeit, Astigmatismus, mäßig verminderte Sehschärfe, normale Fundi und Protanopie. Das ERG zeigte anomale Zapfenreaktionen, aber normale Stäbchenreaktionen. Psychophysikalische Tests zeigten eine selektive Beeinträchtigung Langwellen-(L)-Zapfen in Kombination mit gut erhaltener Mittel-(M)- und Kurzwellen-(S)- Zapfenfunktion.
In der vorherigen Studie stellten Michaelides et al. (2005) [9] fest, dass in einer der BED-Familien die bekannte durch das Exon 4, codierte schädliche Cys203Arg-Substitution abgesondert wurde. Diese Mutation könnte daher für die beobachtete Zapfenfunktionsstörung verantwortlich sein, wenn sie in einer Untergruppe von Zapfen bei den betroffenen Personen exprimiert ist. Im Gegensatz dazu konnten sie in den übrigen untersuchten Familien keine schädlichen Punktmutationen in den L- und M-Opsin-Genen finden. Im Jahr 2014 wurden in ihrer neuen Studie nicht nur die Opsin-Gene in diesen Familien erneut untersucht, sondern die Untersuchungen auf die ursprünglichen BED-Familien, die Familie aus Minnesota und auf 3 plus eine neue britische Familie ausgeweitet; in allen Fällen ist eine seltene Kombination (Haplotyp) von Aminosäuren vorhanden, entweder LVAVA oder LIAVA, spezifiziert durch die Codonen 153, 171, 174, 178, und 180 des Exons 3 des L-Opsin-Gens, eine Kombination, die bisher in nicht mehr als 300 L- oder M-Opsin-Genen oder in separaten Studien der L- und M-Gene in einer großen Anzahl von Dichromaten bekannt war.
Diese zwei Haplotypen scheinen daher auf Personen beschränkt zu sein, bei denen entweder BED oder ähnliche Zapfenfunktionsstörungen diagnostiziert wurden.
Die Sequenz- und Genreihenanalyse der L- und M-Gene in den verschiedenen untersuchten BED-Familien zeigt, dass in vielen Fällen mehrere Gene innerhalb des Arrays vorhanden sind. Bei Untersuchungen kodiert das vorgeschaltete oder proximale Gen jedoch den einen oder anderen Variantenhaplotypen. Eine frühere Studie zur Blauzapfen-Monochromasie implizierte, dass diese Variantenkombinationen von Aminosäuren nicht funktionelle Sehpigmente hervorrufen; wenn dies auch für BED zutrifft, könnte die Dichromasie durch die Expression eines nicht funktionellen Sehpigments aus dem proximalen Opsin-Gen im Array entstehen, wobei die Art der Dichromasie von den spektralen Eigenschaften des funktionellen Sehpigments abhängt, die durch das zweite Gen im Array ausgedrückt wird.
Ein konstantes Merkmal in allen BED-Familien ist Kurzsichtigkeit. Die genetische Grundlage für Kurzsichtigkeit ist Gegenstand mehrerer neuerer Studien. Es wurden eine Reihe genetischer Loci für hohe und mittlere Kurzsichtigkeit identifiziert, hauptsächlich aus Studien mit Familienstämmen, und viele von ihnen wurden in einer internationalen, kollaborativen Studie über das gesamte Genom der Kurzsichtigkeit repliziert. Dazu gehört der Locus MYP1 (OMIM-Nummer 310460), welcher der Spitze des X-Chromosoms bei Xq28 zugeordnet ist. Dieser wurde erstmals in der ursprünglichen BED-Familie identifiziert. Eine kürzlich in China durchgeführte Studie bestätigte seine Position im Chromosom Xq28 innerhalb eines Abstands von 6,1 cM zwischen dem Marker DXS8069 und Xqter. In der Studie konnten jedoch in vier Kandidatengenen keine Mutationen gefunden werden, die aufgrund der Expression im Auge und der Abwesenheit anderer Krankheitsassoziationen ausgewählt wurden; die Opsin-Gene, die ebenfalls dieser Region des X-Chromosoms zugeordnet sind, wurden nicht untersucht. Es scheint heute bekannt zu sein, dass die Opsin-Genvariation sowohl für die Funktionsstörung des Zapfens als auch für die Farbwahrnehmungsstörung verantwortlich ist. Da Kurzsichtigkeit ein integraler Bestandteil von BED ist, muss diese auch durch das dysfunktionale Zapfen-Sehpigment und die Unterbrechung des normalen Zapfenmosaiks entstehen. Wie dies jedoch zu Kurzsichtigkeit führt, bleibt unklar. An dieser Stelle wird darauf hingewiesen werden, dass Kurzsichtigkeit auch in der Blauzapfen-Monochromasie ein häufiges Merkmal ist.
Molekulargenetik
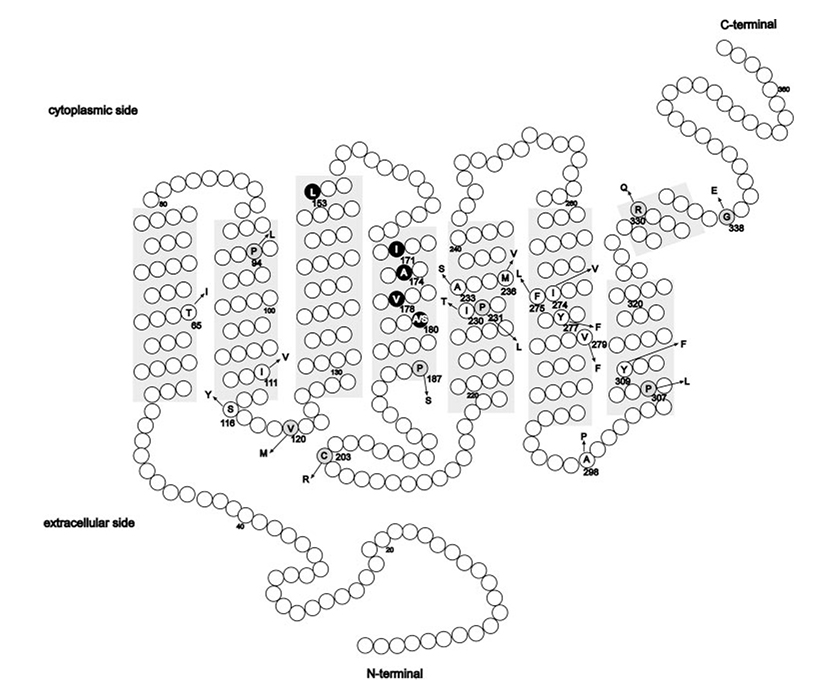
Mutationen in den OPN1LW- und OPN1MW-Zapfen-Opsin-Genen liegen einem Spektrum von Zapfen-Fotorezeptorfehlern von stationärem Verlust der Farbwahrnehmung bis hin zu fortschreitender Netzhautdegeneration zugrunde.
Mutationen können in drei Klassen unterteilt werden: Deletionen der Locus-Kontrollregion (LCR); Missense-Mutation (S. Cys203Arg) in einem L/M-Hybridgen und anderen Punktmutationen; und Exon-3-Single-Nucleotide-Polymorphisms (SNP) tauschen Haplotypen in einem ansonsten normalen Gen-Array aus. Erfahren Sie mehr über ursächliche Mutationen auf dem Opsin-Gen-Array
Bei allen Mutationskategorien wurde eine mittlere bis hohe Kurzsichtigkeit beobachtet. Personen mit LCR-Deletionen oder p.Cys203Arg-Mutationen hatten mit höherer Wahrscheinlichkeit Nystagmus und Sehstörungen, mit langsamer Krankheitsprogression [19]. Es wurden drei krankheitsassoziierte Exon-3-SNP-Haplotypen identifiziert, die LIAVA, LVAVA oder MIAVA kodieren. Patienten mit der letztgenannten Mutation – SNP-Haplotyp – hatten weniger häufig Nystagmus, zeigten aber häufiger eine Progression. Es wurde gezeigt, dass der Haplotyp LIAVA zu einem Exon-3-Skipping führt. Die Haplotypen LVAVA und MIAVA führen ebenfalls zu fehlerhaftem Spleißen mit einem Restgehalt an korrekt gespleißtem Zapfen-Opsin. Das OPN1LW/OPN1MW:c.532A>G SNP, das allen drei krankheitsassoziierten Haplotypen (LIAVA; LVAVA; MIAVA) gemeinsam ist, scheint hauptsächlich für diesen Mutationsmechanismus (Spleißen) verantwortlich zu sein.
Das Auftreten der Bornholm-Augenkrankheit (BED) oder von Blauzapfen-Monochromasie (BCM) hängt davon ab, wie viele Kopien (nur 1 für BED oder mehr für BCM) der Patient in seinem Opsin-Gencluster und welchen Exon-3-SNP-Haplotyp er hat.
Patienten mit einem einzelnen roten oder rot-grünen Hybridgen und einem schädlichen Haplotyp wie LIAVA oder LVAVA weisen den BCM-Phänotyp auf. LIAVA/LVAVA und andere Genotypen führen zu einem falschen Spleißen der mRNA von Opsin und einem nicht funktionellen Opsin. Buena-Atienza et al. [17]
Patienten, die zwei oder mehr Kopien im Cluster haben, und nur das erste L-Gen im Cluster oder bei denen nur das erste M-Gen den schädlichen SNP-Haplotyp hat, weisen den Bornholm-Augenkrankheits-Phänotyp auf. Man könnte sie als Protanope (oder Deuteranope, bei betroffenem zweitem Gen) betrachten, aber bei dieser Art von Mutation (SNP) scheint der Phänotyp nicht nur Farbenblindheit zu sein, da es sich um einen Phänotyp mit (oft) progressiver Zapfendysfunktion/-dystrophie und hoher Kurzsichtigkeit und Dichromasie handelt. Weitere klinische Studien solcher Patienten sind erforderlich, um die Krankheit und den Krankheitsverlauf vollständig zu verstehen.
Publikationen
[1] Bartsocas CS, Kastrantas AD,’X-linked form of myopia’. Hum. Hered.(1981) 31: 199-200.PDF
[2] Haim M, Fledelius HC, Skarsholm D,’X-linked myopia in a Danish family’. Acta Ophthalmol (Copenh). (1988) 66:450-456. PubMed
[3] Schwartz M, Haim M, Skarsholm D,’X-linked myopia: Bornholm eye disease linkage to DNA markers on the distal part of Xq’. Clin Genet. (1990) 38:281-286.PubMed
[4] Mantyjarvi M, Katajakunnas M, Vanttinen S,’High myopia with cone dysfunction’. Acta Ophthalmol (Copenh). 1991;69:155-161.PubMed
[5] Winderickx J, Sanocki E, Lindsey D, Teller DY, Motulsky AG, Deeb SS,’Defective colour vision associated with missense mutation in the human green visual pigment gene’. Nat Genet. 1992;1:251-266.
[6] Hanna MC, Platts JT, Kirkness EF,’Identification of a gene within the tandem array of red and green color pigment genes’. Genomics (1997) 43: 384-386. PubMed
[7] Johnson S, Halford S, Mollon JD, Moore AT, Hunt DM,’Molecular basis of cone dystrophy associated with protanopia’. Invest Ophthalmol Vis Sci. 2001;42(suppl): 3442.Nuffield
[8] Young TL, Deeb SS, Ronan SM, Dewan AT, Alvear AB, Scavello GS, Paluru PC, Brott MS, Hayashi T, Holleschau AM, Benegas N, Schwartz M, Atwood LD, Oetting WS, Rosenberg T, Motulsky AG, King RA,’X-Linked High Myopia Associated with Cone Dysfunction’. Ophthalmic Molecular Genetics (2004) Vol.22 897-908.PubMed
[9] Michaelides M, Johnson S, Bradshaw K, Holder GE, Simunovic MP, Mollon JD, Moore AT, Hunt DM,’X-linked cone dysfuncion syndrome with myopia and protanopia’. Ophthalmology 112 1448-1454 (2005).PubMed
[10] Chen Y-T, Iseli C, Venditti CA, Old LJ, Simpson AJG, Jongeneel CV,’Identification of a new cancer/testis gene family, CT47, among expressed multicopy genes on the human X chromosome’. Genes Chromosomes Cancer 45: 392-400, 2006.
[11] Metlapally R, Michaelides M, Bulusu A, Li Y-J, Schwartz M, Rosenberg T, Hunt DM, Moore AT, Züchner S, Rickman CB and Young TL, ‘Evaluation of the X-Linked High-Grade Myopia Locus (MYP1) with Cone Dysfunction and Color Vision Deficiencies’. Invest. Ophthalmol. Vis. Sci. (2009) April; 50(4) 1552–1558. doi:10.1167/iovs.08-2455 PDF
[12] Ratnamala U, Lyle R, Raval R, Singh R, Vishnupriya S, Himabindu P, Rao VV, Aggarwal S, Paluru P, Bartoloni L, Young TL, Paoloni-Giacobino A, Morris MA, Nath S, Antonarakis SE, Radhakrishna U,’Refinement of the X-linked nonsyndromic high-grade myopia locus (MYP1) on Xq28 and exclusion of thirteen known positional candidate genes by direct sequencing’. Invest. Ophthal. Vis. Sci. (2011) 52: 7909.PubMed
[13] Gardner JC, Liew G, Quan Y-H, Ermetal B, Ueyama H, Davidson AE, Schwarz N, Kanuga N, Chana R, Maher ER, Webster AR, Holder GE, Robson AG, Cheetham ME, Liebelt J, Ruddle JB, Moore AT, Michaelides M and Hardcastle AJ,’Three Different Cone Opsin Gene Array Mutational Mechanisms; Genotype-Phenotype Correlation and Functional Investigation of Cone Opsin Variants’. Hum Mutat. (2014) 35(11):1354-62. doi: 10.1002/humu.22679. PubMed
[14] McClements ME, Neitz M, Moore AT, Hunt DM,’Bornholm Eye Disease Arises From a Specific Combination of Amino Acid Changes Encoded by Exon 3 of the L/M Cone Opsin Gene’. ARVO Annual Meeting Abstract (2010) Volume 51, Issue 13.
[15] McClements M, Davies WIL, Michaelides M, Young T, Neitz M, MacLaren RE, Moore AT and Hunt DM, ‘Variations in opsin coding sequences cause X-linked cone dysfunction syndrome with myopia and dichromacy’.
[16] Ueyama H, Muraki-Oda S, Yamade S, Tanabe S, Yamashita T, Shichida Y, Ogita H,’Unique haplotype in exon 3 of cone opsin mRNA affects splicing of its precursor, leading to congenital color vision defect’. Biochem Biophys Res Commun. (2012) 424(1):152-7. doi: 10.1016/j.bbrc.2012.06.094.PubMed
[17] Buena-Atienza E, Rüther K, Baumann B, Bergholz R, Birch D, De Baere E, Dollfus H, Greally MT, Gustavsson P, Hamel CP, Heckenlively JR, Leroy BP, Plomp AS, Pott JWR, Rose K, Rosenberg T, Stark Z, Verheij JBGM, Weleber R, Zobor D, Weisschuh N, Kohl S, Wissinger B,’De novo intrachromosomal gene conversion from OPN1MW to OPN1LW in the male germline results in Blue Cone Monochromacy’. Scientific Reports, 6, [28253]. DOI: 10.1038/srep28253 Nature
[18] Patterson EJ, Wilk M, Langlo CS, Kasilian M, Ring M, Hufnagel RB, Dubis AM, Tee JJ, Kalitzeos A, Gardner JC, Ahmed ZM, Sisk RA, Larsen M, Sjoberg S, Connor TB, Dubra A, Neitz J, Hardcastle AJ, Neitz M, Michaelides M, Carroll J,’Cone Photoreceptor Structure in Patients With X-Linked Cone Dysfunction and Red-Green Color Vision Deficiency’. Invest Ophthalmol Vis Sci. 2016 Jul 1;57(8):3853-63. doi: 10.1167/iovs.16-19608. PubMed
[19] Sumaroka A, Garafalo AV, Cideciyan AV, Charng J, Roman AJ, Choi W, Saxena S, Aksianiuk V, Kohl S, Wissinger B, Jacobson SG. (2018) “Blue cone monochromacy caused by the C203R missense mutation or large deletion mutations”. Invest Ophthalmol Vis Sci. 2018 Dec 3;59(15):5762-72. doi:https://doi.org/10.1167/iovs.18-25280
